Understanding why the brain ages differently—and more slowly—than other organs has been one of neuroscience's enduring puzzles. This study reframes that question dramatically: the brain may not initiate its own decline but instead receive toxic inflammatory signals from the periphery, suggesting that systemic inflammation control could be a lever for protecting the aging brain before neurological symptoms ever appear.
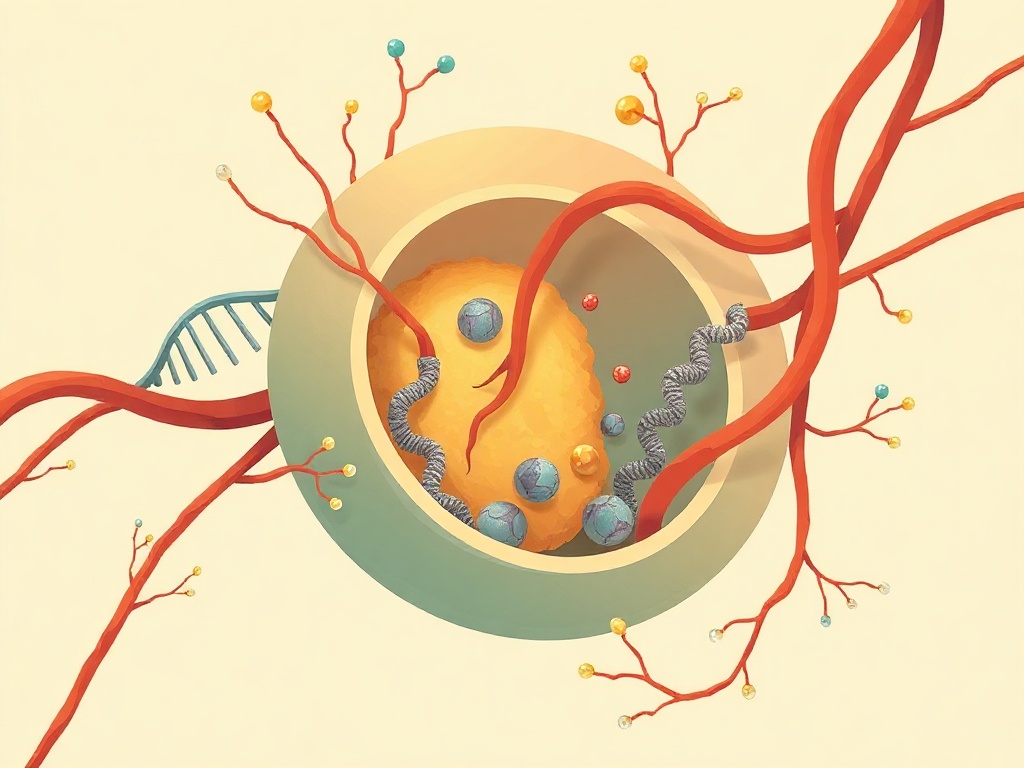
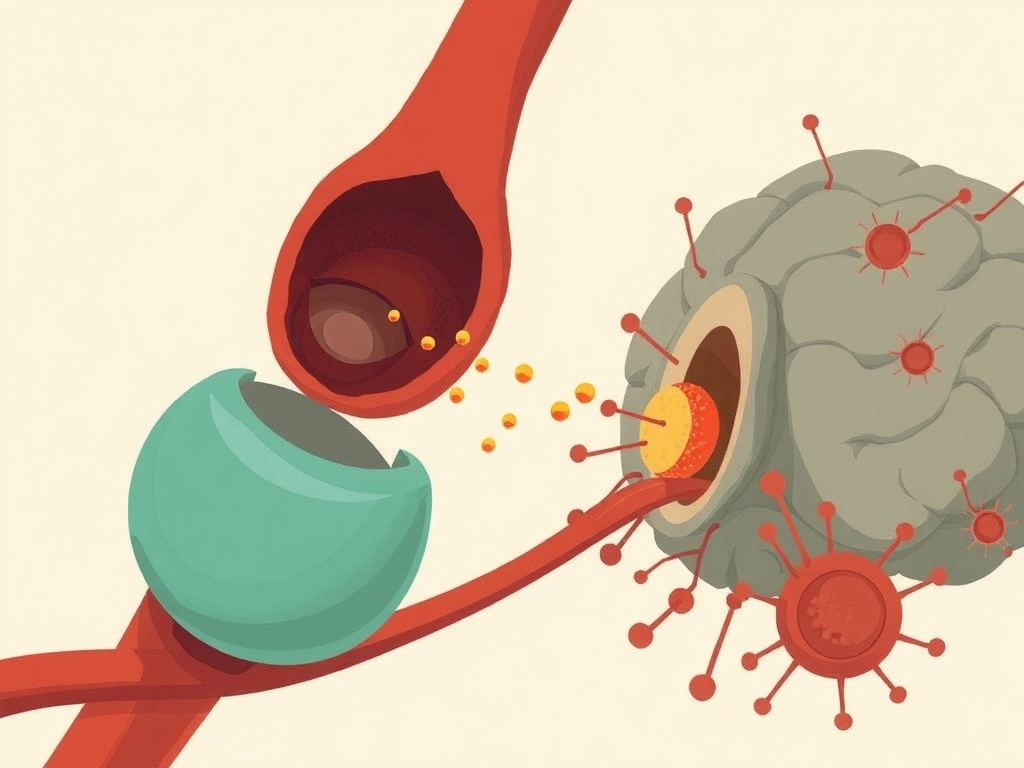
Working across Parkinson's disease patient samples and a genetically engineered mouse model carrying LRRK2 gain-of-function (GoF) mutations—among the most significant known genetic risk factors for Parkinson's—the researchers traced a mechanistic cascade from cellular aging outward to the brain. As cells age or carry LRRK2GoF mutations, their endolysosomal compartments deteriorate, allowing self-DNA to accumulate in the cytosol. This displaced DNA is then packaged into extracellular vesicles (EVs) and released into circulation. These DNA-laden EVs activate the cGAS-STING innate immune pathway both within originating cells and in recipient cells at distant sites. The resulting inflammatory signaling eventually compromises the blood-brain barrier, enabling neuroinflammation and dopaminergic neuron loss characteristic of Parkinson's disease.
This work sits at the intersection of two rapidly converging fields: inflammaging—the chronic low-grade inflammation increasingly recognized as a driver of age-related disease—and extracellular vesicle biology. The cGAS-STING pathway has attracted intense pharmacological interest across oncology and autoimmunity, and multiple STING antagonists are currently in early clinical development. The finding that LRRK2GoF accelerates a systemic aging phenotype rather than causing purely local brain pathology is conceptually important: it implies Parkinson's may be diagnosable and potentially interceptable at a peripheral inflammatory stage, well before neurodegeneration is detectable. Key limitations include the reliance on a gain-of-function genetic model that represents only a subset of Parkinson's cases, and the mechanistic chain—while compelling in mice—requires prospective validation in human cohorts. Still, this is a genuinely paradigm-shifting framing of Parkinson's as accelerated systemic inflammaging, not merely a brain disease.